On March 15, 2019 the FDA issued a statement indicating they are bolstering their efforts to better evaluate the safety profile and biocompatibility of materials used in medical devices. This move is hot on the heels of the ICIJ Implant Files and indicates the agency will conduct a public hearing later this year to “help gain a broader understanding of nitinol and other metals in devices…and the potential risk for certain patients to have ‘hypersensitivity’ or exaggerated immune and inflammatory reactions to these metals.”
There is a ton to digest in this announcement, and we can all agree that it is a step in the right direction. However, there are still a number of statements which can reasonably be argued as false and misleading.
1) The FDA statement that adverse events only happen to a very small percentage of patients is not unlike a house built upon sand. The agency has no numerator or denominator upon which to make that claim, and if you examine AE reports along with patient-centered social media groups, you will find the numbers of harmed patients is quite large. The FDA does not know how many people have been implanted with a specific device and thus do not know exact percentages of people experiencing issues after implantation. Even if they had that information, the FDA hasn’t established any explicit guidelines to say how many injured patients are too many or qualify as something other than “small percentage harmed.”
2) A 2009 GAO report faulted the FDA for its handling, reading, tracking, and response to adverse event reports – including deaths. We have not seen much evidence that this has changed. With the known under-reporting, consolidated summary reports from industry, and the KHN investigation revealing hidden reports, we do not think they have much ground to stand upon here. It seems the FDA still struggles heavily with post-market surveillance and adverse-event reports.
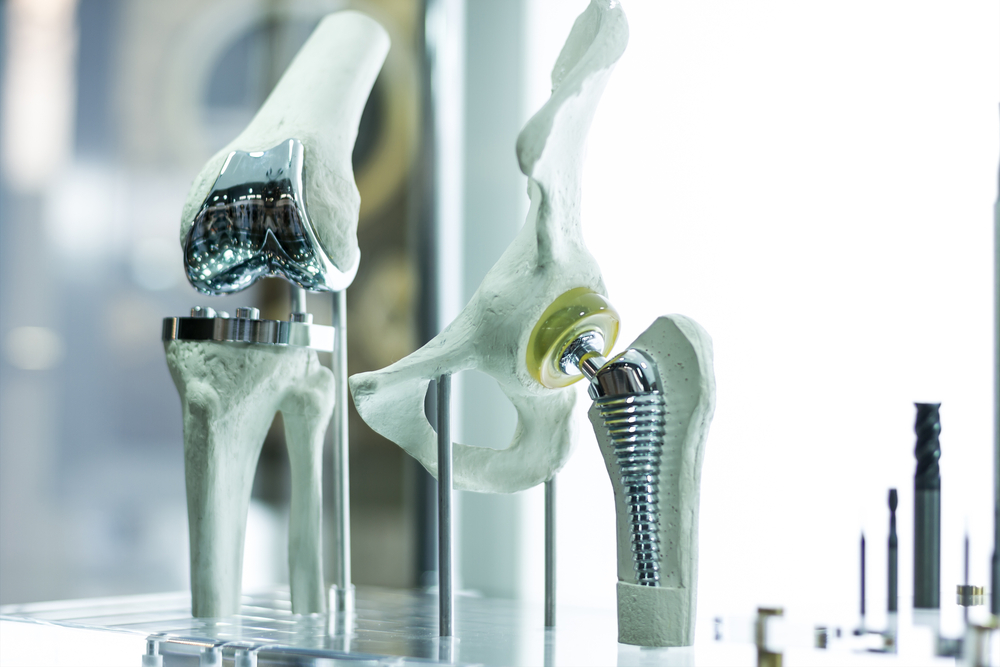
3) Gottlieb claims they do careful reviews of materials for biocompatibility and more, but the number of devices which have lengthy histories of causing harm are telling a very different story. During the premarket approval meeting for Essure, the FDA panel was NOT provided the biocompatibility testing information. It has always been presented to the FDA and to patients in a comparative way to heart stents. However, experts who spoke at the 2015 FDA hearing on Essure explained the differences. Heart stents used in arteries may be easier to detox through the bloodstream than a device such as Essure, where the metals are encapsulated by surrounding mucosal tissues. As we saw with Dr. Tower in The Bleeding Edge with his metal-on-metal hip implant, a metal sludge accumulated in the surrounding tissue when he had a surgical revision to replace his failing implant.
If biocompatibility had been carefully reviewed, the issue of corrosion with the use of dissimilar metals in devices such as in metal-on-metal hips and Essure should have been discovered well in advance of market use. This corrosion issue, especially at locations of mechanical function such as joints, is a topic of basic high school science, yet the device engineers and FDA scientists all appear to have glossed over this important safety issue when approving these devices.
4) Gottlieb focused on the nitinol in Essure as the possible cause of the inflammatory response. While hypersensitivity to metals and allergic reactions are one possible outcome, he completely avoided mentioning the reported mechanism of action of Essure: the PET fibers (polyethylene terephthalate). The inner stainless steel coil of Essure, which also contains nickel, is wrapped and woven with the PET fibers. The PURPOSE of the PET is to cause a chronic inflammatory response to initiate scar tissue growth, thus occluding the fallopian tubes. What happens if and when that reaction continues or if the device migrates to a different area of the body OR if the device is cut/pulled/broken/fractured during improper removal, subsequently exposing the abdomen to these tiny untraceable hair like fibers?
5) While we appreciate Gottlieb’s candor about the problems, we are left with many unanswered questions regarding patient representation in the work that is ahead and the realities of both pre and post-market practices, especially within the 510(k) pathway. Fundamentally, the work being proposed is all science homework that should have been done prior to allowing the devices to go to market. In that regard, it feels like the FDA is trying to scramble after having closed the barn doors after the horses had already escaped.
6) Further, why isn’t polypropylene mesh mentioned as part of this investigation? Is it the same reason the FDA decided to focus on metals in devices while excluding talk of PET in Essure? Plastic materials in implants also have a long history of being questionable. Suppliers of the raw materials began to withhold product from device companies due to being dragged into lawsuits involving medical devices containing those raw materials. Most plastic materials even come with a warning to NOT be implanted permanently into the human body. The fact that enough legal complaints occurred for the suppliers to withhold materials alone should be a HUGE red flag to the FDA! However, greed won out and lobbyists convinced Congress to introduce and pass the Biomaterials Access Assurance Act of 1998, which left clinical testing under control of the device manufacturers along with self-reporting adverse events.
At the end of the day, all of our misgivings about this still point to regulatory capture of the FDA by the very industry it is supposed to police:
- number of devices cleared for market without sufficient scientific data of safety & effectiveness
- the failure of the FDA to enforce post-market studies
- the free flow of large sums of money to Congress from industry
- the long delays and reluctance of the FDA to ban or recall harmful devices.
This poses a very significant threat to the health of the American public; we rely on accurate, non-biased information and a reasonable assurance of safety for products regulated by the FDA, seemingly to our own detriment.F